Sepsis - Definition
The word sepsis is derived from the Greek, sêpsis meaning “decay or “to rotten.” During the last century, this term was used to describe a broad range of systemic infectious disease states independent of the severity of the illness. The absence of precise terminology to describe protean manifestations and the degree of disease hampered clinical trial design and the development of management strategies for sepsis. Clinical experience suggested that the presence of shock and the development of organ failure syndromes were key factors in determining outcome from serious infections yet these characteristics were not delineated by the term sepsis.
To address the need for more precise terminology, a consensus conference standardized the definitions used to describe severe infections and their sequelae (Table 1). These definitions recognized that following the development of an infection or after sustaining an injury (i.e. trauma or pancreatitis), a systemic inflammatory response syndrome (SIRS) occurs that can be manifested by fever, tachycardia, tachypnea and leukocytosis. “Sepsis” was defined as the invasion of sterile tissue by one or more microbial pathogens with a resultant systemic inflammatory response. It is important to note, however, that in 70% of cases of sepsis, the offending pathogen could not be identified although infection seemed to be the only plausible initiating agent. The severity of sepsis was delineated based on responses to fluid resuscitation and the presence of new organ failure syndromes (e.g. coagulopathy or renal insufficiency). “Severe sepsis” compared to “sepsis” was characterized by a more severe systemic inflammatory response to infection resulting in hypotension and organ hypoperfusion (e.g. renal failure, impaired mentation or hypoxemia). “Septic shock” was the more grave consequence of an infection manifested by persistent hypotension despite adequate fluid resuscitation and organ dysfunction.
When evaluated prospectively these definitions describe a continuum from systemic inflammatory responses, to sepsis (26% of patients with evidence of systemic inflammatory response), to severe sepsis (18% of septic patients) and ultimately septic shock (4% of severe septic patients). The risk of death increased incrementally with the severity of illness (systemic inflammation 7%, sepsis, 16%, severe sepsis 20% and septic shock 46%). This hierarchical model of the severity of infection and the clinical manifestations of the host response to infection has provided a pragmatic to tool to compare clinical manifestations of infectious syndromes.
Table 1. Definitions of Sepsis and Organ Failure
| Definition |
Crude mortality |
|
|
Infection |
Pathologic process caused by the invasion of normally sterile tissue or fluid or body cavity by pathogenic microorganisms |
|
|
Systemic Inflammatory Response Syndrome |
Response to infection or other severe clinical insult manifested by two or more of the following: Temperature: > 38° C or <36° C Heart rate: > 90 beats/min Respiratory rate: > 20 breaths/min or PaCO2 < 32 mm Hg WBC>12,000 cells/mm3, <4,000 cells/mm3 or >10% bands |
7% |
|
Sepsis |
Clinical syndrome defined by the presence of both infection and a systemic inflammatory response |
15% |
|
Severe Sepsis |
Sepsis with organ dysfunction as evidenced by hypotension or hypoperfusion(i.e., hypoxemia, renal failure, change in mental status) |
20% |
|
Septic Shock |
Severe sepsis with hypoperfusion despite adequate fluid resuscitation |
45% |
|
Multiple Organ Dysfunction Syndrome |
Presence of altered organ function in an acutely ill patient such that homeostasis cannot be maintained without intervention |
|
Epidemiology
In the United States sepsis causes more than 200,000 deaths annually and is the second most common infectious cause of death in the United States after pneumonia. The crude mortality rate is 15%, but increases to 20 and 45% for severe sepsis and septic shock respectively (Table 1). Recent epidemiologic data suggests that the incidence of sepsis is increasing at a rate of 8.7% annually. A number of factors are likely to be contributing to this increase: improved recognition and reporting; increased age of the general population; increased numbers of patients at risk for sepsis because of compromised immunity (i.e. HIV infection, therapies for malignancies or organ transplant recipients); and increased use of indwelling devices (e.g. vascular access devices) as well as the emergence of newly resistant pathogens in the community. The risk of sepsis is greatest in the very young and the elderly. Among pediatric patients, sepsis is more common in infants less than one year of age and affects boys more than girls. The incidence of sepsis is higher in men than women and occurs more frequently in minorities.
Over the last fifty years, there has been a change in the predominant organisms causing sepsis. In the early 1960s, gram-negative bacilli were the most common cause of bacteremia and sepsis whereas in the last two decades, gram-positive cocci and yeasts have emerged as the major microbial pathogens in sepsis. Patients who survive an episode of sepsis have an increased risk of dying during the subsequent five years when compared to controls. Whether this is a direct result of the episode of sepsis, or is a marker of co-morbid conditions that predisposed to the development of sepsis is not known.
Pathogenesis
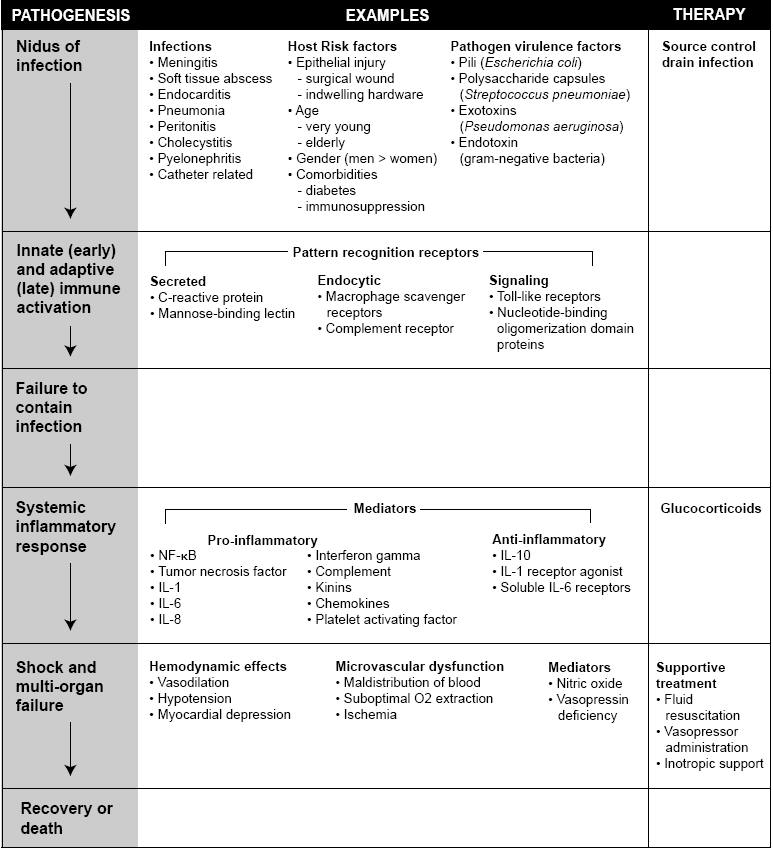
Sepsis results from a systemic immune response with multiple inflammatory mediators activated by microbial invasion of sterile tissue or the blood stream. These inflammatory responses are necessary to survive an infection, but may also result in host tissue injury (i.e. acute lung injury, hypotension or acute renal failure). Sepsis includes a broad range of syndromes. The clinical picture of a child in the throes of purpura fulminans and an elderly patient with urosepsis are strikingly different, yet there are aspects of host immune response and pathologic mechanisms that are common to both of these septic patients (Figure 1).
Figure 1: Pathogenesis and therapy of sepsis and septic shock. Outline of the sequence of events that occur during the pathogenesis of sepsis and septic shock including examples of mediators and standard therapy.
Innate Immunity: Detection and Early Response to Infection
Infection can initiate a cascade of inflammatory events that results in sepsis. Characteristics of the host’s immunologic integrity (i.e. age, comorbidities, or indwelling hardware) and the pathogen’s virulence (i.e. factors such as pili, polysaccharide capsules and exotoxins) influence the severity of the syndrome and the patient’s outcome. Host survival requires the ability to identify, contain and eradicate a microbial invader. Innate immunity represents the immediate nonpathogen specific host response to infection. Adaptive immunity, on the other hand, is a less rapid (occurring over three to five days) but more precise response to an infection requiring the clonal expansion of lymphocytes and the production of antibody to a specific pathogen.
The innate immune system engages pathogens via pattern recognition receptors (PRR). These receptors are genetically predetermined and are believed to have evolved through natural selection. The PRRs bind to molecular structures termed pathogen-associated molecular patterns (PAMPs) which are shared across microbial species (e.g. endotoxin or peptidoglycan). Host pattern recognition receptors can be divided into three groups: secreted, endocytic and signaling. Mannose-binding lectin and C-reactive protein, are prototypical secreted PRRs that bind to microbial cell membrane components leading to complement activation. Endocytic PRRs such as macrophage mannose receptor are located on the surface of phagocytes and facilitate the phagocytosis of pathogens. Signaling PRRs, include Toll-like receptors (TLR) and the nucleotide-binding oligomerization (NOD) proteins, NOD1 and NOD2. Ten human TLRs have been discovered on the surface of various cells including phagocytes. Unlike TLRs which are cell membrane bound, NOD1 and NOD2 are intracellular proteins found in epithelial, monocytes, dendritic cells and granulocytes where they bind to bacterial peptidoglycan moieties. The diversity of ligands for TLRs and NODs permit the detection and subsequent response to a broad range of infections. And both TLRs and NODs have attracted interest as potential targets for therapeutic blockade.
Cytokines and Other Mediators: Signal Amplification and Coordination of the Immune Response
Both TLR and NOD pathways lead to the activation of nuclear factor kappa binding (NF-κB) transcription factor and amplification of the initiating signal via the production of cytokines. In general, cytokines are signaling molecules which can be classified as either pro or anti-inflammatory. Activation of NF-κB leads to the synthesis and release of pro-inflammatory cytokines tumor necrosis factor (TNF), and IL-1 by cells of the innate immune system including monocytes and macrophages. These signaling molecules augment a series of local and systemic inflammatory responses and engage various components of the immune and coagulation systems including: acting on the hypothalamus to induce fever; stimulation of bone marrow to release neutrophils; activation of endothelium to promote extravasation of phagocytes and complement to the site of infection; triggering of hepatocytes to synthesize C-reactive protein which binds cell wall components of bacteria, fungi and parasites; and the promotion of platelet-endothelial adhesion with clot formation. In addition to IL-1 and TNF other prominent pro-inflammatory cytokines and immune modulators involved in the immune response to infection include IL-8, IL-12 and interferon gamma. Absolute pro-inflammatory cytokine deficiencies can predispose the host to overwhelming infection and death while excessive amounts can cause damage to the host. Elevated levels of pro-inflammatory cytokines are also associated with increased mortality and experimental infusions of TNF produce a clinical syndrome identical to sepsis which can be reversed with antibody therapy specific to TNF. Thus, the systemic effects of cytokines including vasodilatory shock and disseminated intravascular coagulation have been implicated in the pathogenesis of sepsis. Nonetheless, while clinical trials aimed at improving survival in septic shock have targeted these pro-inflammatory cytokines, no trial inhibiting a single cytokine has shown clinical benefit.
Immunosuppression: Confining Inflammation to the Site of Infection or Predisposing the Host to Sepsis
Paradoxically, infection induces host production of anti-inflammatory mediators. Traditionally, this immunosuppressive response was thought to curb inflammation during the recovery phase of infection. More recent evidence suggests that normally pro and anti-inflammatory signaling pathways are activated simultaneously and serve to concentrate the immune response at the site of insult all the while limiting systemic immune activation. Circulating IL-10 which inhibits phagocyte activation and cortisol which inhibits cytokine synthesis by monocytes are both increased in the setting of infection. Similarly, blood levels of both IL-1 receptor agonist and soluble IL-6 receptors are also elevated with infection thereby limiting the systemic effect of their respective pro-inflammatory ligands. Inflammatory markers in bronchial washings of patients with ARDS with paired blood samples from the same host are consistent with this dichotomous model of inflammation at the site of infection and systemic immunosuppression. Appreciation for the role of anti-inflammatory mechanisms has given rise to the thought that some of the complications of sepsis may in part be a function of an immunosuppressed state (referred to as immune paralysis) in which anti-inflammatory cytokine levels are disproportionately elevated and immune cells either respond poorly to pro-inflammatory stimulation or actually undergoing apoptosis.
Septic Shock and Subsequent Organ Dysfunction
Sepsis may progress to cardiovascular collapse and multiple organ dysfunction that culminate in death or residual organ failure. Vascular endothelial dysfunction is central to the development of the hypotension and end organ injury in sepsis. Nitric oxide (NO), a signaling molecule and potent vasodilator, is produced by nitric oxide synthase (NOS) present in vascular endothelium and smooth muscles cells. In sepsis an inducible isoform of NOS (iNOS) is over expressed in response to pro-inflammatory cytokines. The resultant increase in NO plays a prominent role in the vasodilatation that is a hallmark of septic shock. Administration of NOS inhibitors to septic patients reverses shock, but also increased mortality secondary to widespread tissue ischemia. Vasopressin deficiency which is frequently present in septic patients may contribute to septic shock and organ dysfunction at the vascular endothelial level.
In addition to systemic hypotension, maldistribution of blood flow between organ systems has also been implicated as a contributory factor in sepsis. Microvascular dysfunction secondary to activation of the coagulation system and the formation of micro thrombi further impairs oxygen and nutrient delivery at the cellular level. Even when blood flow is believed to be adequate there is evidence that tissues are unable to optimally extract oxygen. This may in part be due to decreased mitochondrial function.
In addition to the direct effect on the vasculature, sepsis directly impairs cardiac performance which further compromises organ perfusion. The usual hemodynamic profile of septic shock includes high cardiac output, low mean arterial pressure and tachycardia. Left ventricular function is depressed as evidenced by a decrease in the ejection fraction. Patients who survive sepsis maintain stroke volume by undergoing acute ventricular dilation which reverses during their recovery phase. Elevated levels of troponin may be observed in sepsis; however the cause myocardial dysfunction is not ischemia as coronary blood flow is maintained. Circulating immune mediators are likely to contribute to the depressed ventricular function observed in sepsis although no specific agent has been identified.