
Fever and Chest Pain - Cardiac Etiologies
The complaint of fever and chest pain warrants a thorough consideration of
cardiac causes. Symptoms such as dyspnea on exertion, orthopnea, and
palpitations should be actively identified. Knowledge of a recent viral illness,
surgical procedure or intravenous drug use may be helpful in identifying an
inciting event. Likewise, risk factors such as a prosthetic valve or previous
episodes of endocarditis should be identified. The physical examination may
yield a wealth of clues such as a new heart murmur, pericardial rub, petechiae,
splenomegaly or peripheral stigmata of embolic disease (e.g. Janeway lesions,
Osler nodes, splinter hemorrhages). Electrocardiography should be obtained on
all patients with chest pain, regardless of the presence or absence of fever,
and may show evidence of myocardial involvement. While most laboratory studies
are nonspecific, cardiac biomarkers may reveal active, ongoing myocardial
injury. Blood cultures are indispensable to the isolation of causative
microorganisms. Echocardiography may be an important diagnostic tool for guiding
treatment and the potential need for surgical intervention. Fever and chest pain
may occasionally present together in the patient with acute coronary syndrome or
aortic dissection. While this would be unusual, the astute physician should
always consider these catastrophic cardiac diseases in any patient who presents
with chest pain. However, the bulk of the evaluation should focus on the three
classic cardiovascular causes of fever and chest pain: pericarditis, myocarditis
and infective endocarditis (see Table 2).
Table 2: Cardiac Etiologies of Chest
Pain and Fever
| |
Risk factors |
Presentation |
Management |
| Pericarditis |
Idiopathic Infection
Uremia
Myocardial infarction
Malignancy
Trauma
Autoimmune disease
|
Sharp, retrosternal pain
Positional
Pericardial friction rub
Signs of tamponade
Leukocytosis
± Cardiac biomarkers
ECG: diffuse ST elevations
|
± Echocardiogram NSAIDs
|
| Myocarditis |
Infection
Drugs/toxins
Autoimmune disease
|
Variable, Asymptomatic
à Chest pain à
Failure
± S3 or S4
+ Cardiac biomarkers if acute ECG: ± ST-T changes
|
Echocardiogram
Endomyocardial biopsy
Diuretics, ACE-inhibitors, Beta-blockers
± inotropes, assist device or heart transplant
|
| Infective endocarditis |
Valvular disease
Intravenous drug use
Prosthetic valve
Surgical procedures
Implanted devices
Hemodialysis |
Variable
Heart murmur
Peripheral stigmata
Leukocytosis
ECG: ± conduction abnormalities |
Blood cultures
Echocardiogram
Antibiotic therapy
Surgical intervention |
Pericarditis
Acute inflammation of the pericardium is known as pericarditis and accounts for
approximately 5% of all causes of chest pain presenting to the ED. Acute
pericarditis may be complicated by the development of a pericardial effusion
that, if significant enough to compress the cardiac chambers, may produce
cardiac tamponade. More than 80% of cases of acute pericarditis are idiopathic
or presumably viral in origin. Among many others, commonly implicated viruses
have included the enteroviruses (Coxsackie A & B), adenovirus, Epstein Barr
virus (EBV), cytomegalovirus (CMV), and parvovirus B19. Given the self-limited
nature of most cases of acute pericarditis, a definitive etiology is often not
pursued. Less common infectious causes of acute pericarditis may include
bacterial, tuberculosis and fungal infection. These are more likely to be seen
in the immunocompromised patient. Autoimmune diseases such as SLE and rheumatoid
arthritis represent some of the most common noninfectious causes of pericarditis.
Uremia, trauma and cardiac surgery are other well-known noninfectious causes.
While primary tumors of the pericardium are rare, metastatic breast and lung
cancer as well as lymphoma may spread to the pericardium, producing large and
often hemorrhagic effusions. Pericarditis may occur within days after an acute
transmural myocardial infarction as a result of contact with inflamed and
healing myocardium. It may also occur weeks to months later as an
autoimmune-mediated response known as Dressler’s syndrome.
Acute pericarditis classically presents with sharp, retrosternal chest pain that
is sudden in onset. It typically worsens with deep inspiration, with coughing,
or when the patient is supine. It may be improved with sitting upright and
leaning forward. The pain may radiate to one or both trapezius ridges, as both
phrenic nerves traverse the anterior pericardium to innervate these muscle
groups. Pain may also be referred to the neck, shoulders, or arms, making it
difficult to differentiate from the pain of pulmonary embolism and myocardial
ischemia or infarction. Patients frequently present with fever, malaise and
myalgias in association with their chest pain, reminiscent of a viral prodrome,
though the former may be absent in the elderly. A pericardial friction rub is
highly specific for pericarditis and can be heard at one time or another in as
many as 85% of cases of acute pericarditis. It is described as a high-pitched
raspy sound best auscultated at the left sternal border with the patient leaning
forward on end expiration.
A pericardial friction rub may be heard throughout the respiratory cycle and
persists even when the patient is asked to hold their breath, distinguishing it
from a pleural rub. The commonly held belief that a pericardial rub results from
the chafing of the two inflamed pericardial layers against one another is likely
inaccurate, as a rub may be heard even in the presence of a large effusion
separating the layers. Tachycardia, hypotension, jugular venous distension and
pulsus paradoxus (a decrease in systolic blood pressure of more than 10 mmHg
with inspiration) are suggestive of cardiac tamponade. While low-grade fevers
are common, a temperature above 38º C is concerning for purulent bacterial
pericarditis.
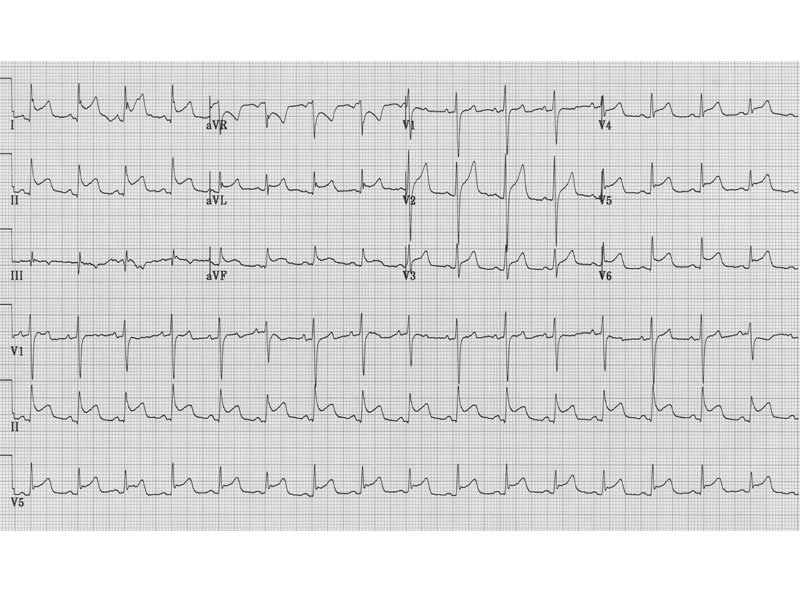
Electrocardiography is often diagnostic for acute pericarditis. While the
pericardium itself is electrically inert, epicardial inflammation from an
overlying pericarditis progresses through four classic stages. Stage 1 is marked
by diffuse, upward concave ST-segment elevations with reciprocal ST-segment
depressions in aVR and V1. PR-segment depression may be seen in most leads with
the exception of aVR and V1 . These changes are seen within the first hours of
initial symptoms and may last up to two weeks before returning to baseline,
defined as stage 2. As inflammation and injury progresses into the second and
third weeks, stage 3 is characterized by diffuse T-wave inversions. Stage 4
marks the resolution of these T-wave inversions, though some may persist
indefinitely. While these four stages are seen less frequently now as a result
of early therapy, the presence of diffuse ST-segment elevations seen in stage 1
still remains a cardinal marker of acute pericarditis. Low voltage QRS complexes
may hint at the presence of a pericardial effusion.
. These changes are seen within the first hours of
initial symptoms and may last up to two weeks before returning to baseline,
defined as stage 2. As inflammation and injury progresses into the second and
third weeks, stage 3 is characterized by diffuse T-wave inversions. Stage 4
marks the resolution of these T-wave inversions, though some may persist
indefinitely. While these four stages are seen less frequently now as a result
of early therapy, the presence of diffuse ST-segment elevations seen in stage 1
still remains a cardinal marker of acute pericarditis. Low voltage QRS complexes
may hint at the presence of a pericardial effusion.
EKG demonstrating typical changes of
pericarditis. Stage 1 is marked by diffuse, upward concave ST-segment
elevations with reciprocal ST-segment depressions in aVR and V1.
Laboratory evaluation in the patient with acute pericarditis may reveal an
elevated white blood cell count, erythrocyte sedimentation rate and serum
C-reactive protein. Renal function should be assessed to exclude uremia as an
etiology. In selected cases, tuberculin skin testing, antinuclear antibody and
rheumatoid factor may aid diagnosis. Serum biomarkers such as creatine kinase
(CK-MB) and serum cardiac troponin I (cTnI) may be elevated in at least a third
of the cases of acute pericarditis and likely reflect superficial myocardial
inflammation and injury. Significant serum cTnI elevations are only seen in the
presence of ST-segment elevation on ECG in pericarditis. Unlike in acute
coronary syndrome, elevated serum cTnI is not associated with a poorer prognosis
in acute pericarditis. However, persistent serum cTnI elevation for more than
two weeks may be suggestive of myocarditis, which does carry a poorer prognosis.
Echocardiography is both appropriate for and frequently obtained in the context
of acute pericarditis to evaluate for the presence of a pericardial effusion
.
While the discovery of an effusion may help solidify a diagnosis of pericarditis,
the absence of one cannot rule it out. In most instances, routine
pericardiocentesis has been demonstrated to have very low diagnostic yield. A
pericardial effusion with evidence of tamponade however is a clear indication to
proceed to pericardiocentesis or surgical drainage. Likewise, an effusion
suspected to be secondary to purulent, tuberculosis or neoplastic pericarditis
warrants sampling of the pericardial fluid to obtain a definitive diagnosis
through bacterial culture, fluid cytology and, if indicated, polymerase chain
reaction (PCR) for tubercle bacilli. In cases of ineffective pericardiocentesis,
tamponade recurs, or the course of pericarditis is prolonged, pericardial biopsy
may be considered.
|
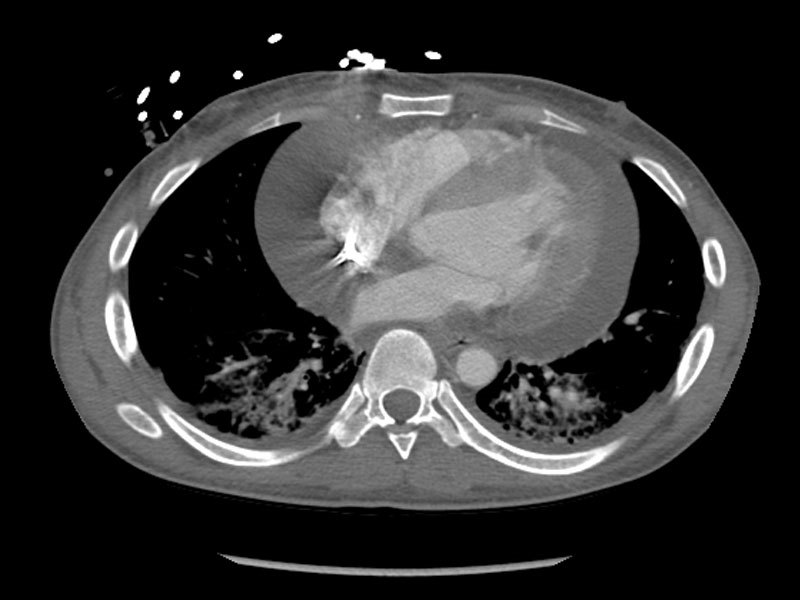
24 year
old with ESRD and shortness of breath. Evaluation revealed a massive
pericardial effusion requiring surgical drainage. |
|
 |
 |
Most cases of acute idiopathic or viral pericarditis respond well to supportive
care and symptom relief with non-steroidal anti-inflammatory drugs (NSAIDs) such
as aspirin, indomethacin and ibuprophen. Aspirin is preferred in patients with
recent myocardial infarction or coronary artery disease, while indomethacin
should be avoided because of its adverse impact on coronary blood flow.
Colchicine has recently been shown to be a safe and effective adjunct to
conventional NSAID therapy in controlling pain and decreasing the risk of
recurrent pericarditis. Systemic steroids should be reserved for recurrent
pericarditis unresponsive to NSAIDs and colchicine or cases of acute
pericarditis linked to an underlying autoimmune or connective tissue disease.
Use of steroids in acute pericarditis has been associated with increased
likelihood of recurrence.
Myocarditis
Myocarditis can be either an acute or chronic inflammatory process of the
myocardium, triggering focal myocyte necrosis and fibrotic deposition. It is a
commonly recognized cause of sudden unexplained cardiac death in young adults.
Depending on the degree of injury, myocarditis may be complicated by dilated
cardiomyopathy and left ventricular dysfunction. In the United States, viral
infections remain the most frequently identified cause of acute myocarditis. As
in pericarditis, enteroviruses (Coxsackie B), adenovirus, EBV, CMV and
parvovirus B19 in addition to other viruses including influenza A, herpes
simplex virus 1 (HSV-1), human herpesvirus 6 (HHV-6) and the human
immunodeficiency virus (HIV) have been implicated in this disease process.
Bacterial and fungal infections make up a small minority of the remaining
infectious causes. Worldwide, protozoal infection with Trypanosoma cruzi, better
known as Chagas’ disease, remains a predominant cause of acute myocarditis and
dilated cardiomyopathy. Noninfectious causes include toxins such as
anthracyclines (doxorubicin) and cocaine as well as hypersensitivity reactions
to tricyclic antidepressants, antibiotics (penicillins, sulfonamides) and
antipsychotics (clozapine). Sarcoidosis, scleroderma and SLE have also been
implicated. Recently, an association has been drawn between smallpox vaccination
and increased incidence of myocarditis in military personnel after widespread
inoculations.
The clinical presentation of myocarditis is highly variable. While some patients
may be completely asymptomatic, others present acutely ill with fever, chest
pain, myalgias, arthralgias, exertional dypnea, palpitations and syncope. In
many cases, these symptoms may be preceded by a nonspecific viral prodrome of
respiratory and gastrointestinal complaints as well as fever, malaise and
headache. Some may present with symptoms consistent with acutely decompensated
heart failure and hemodynamic collapse, suggesting progression to cardiomyopathy.
Auscultation of the chest might reveal a third or fourth heart sound, a new
heart murmur, or evidence of pulmonary congestion, all suggestive of heart
failure. A drug rash might point towards a hypersensitivity reaction as a
possible etiology.
Electrocardiographic changes frequently seen in myocarditis are consistent with
acute injury or ischemia and may in many instances be mistaken for acute
myocardial infarction. ST-segment elevation and depression, T-wave inversions
and pathologic Q-waves have all been seen in cases of biopsy-proven myocarditis
where myocardial infarction was initially suspected and coronary angiography was
subsequently normal. In many of these cases, patients were not only young but
had a dearth of coronary risk factors and had yet presented with symptomatology
and electrocardiographic changes concerning for myocardial ischemia or infarct.
Ventricular arrhythmias and heart block may also manifest with myocarditis and
cardiomyopathy.
The serum cardiac biomarker, cardiac troponin I, is reliably elevated in
patients with myocarditis early on (within one month) after initial onset of
symptoms and is indicative of acute myocyte necrosis. Considered superior to
CK-MB, CTnI may however have returned to normal in patients presenting several
months after initial myocardial injury.
Echocardiography may demonstrate either global ventricular dysfunction or
regional wall motion abnormalities consistent with a nonspecific cardiomyopathy
in patients with signs of heart failure. For patients without these signs, the
echocardiogram may be normal. Evolving noninvasive diagnostic strategies such as
antimyosin scintigraphy and gadolinium-enhanced cardiac magnetic resonance
imaging are increasingly useful tools for differentiating acute myocarditis from
myocardial infarction.
Definitive diagnosis of myocarditis rests with endomyocardial biopsy. Previously
considered to have low sensitivity as a result of the need for multiple biopsies
to obtain a diagnostic result, the yield for biopsy has significantly improved
with the advent of PCR for specific viral genomes. Immunohistochemical assays
for the anti-heart autoantibodies have also helped identify cases of
autoimmune-mediated myocarditis.
The treatment of acute myocarditis remains supportive. Hemodynamic optimization
of heart failure is paramount and should proceed with diuretics to lower
ventricular filling pressures, angiotensin-converting enzyme inhibitors to
reduce vascular resistance, and eventually a beta blocker. In severe cases,
intravenous inotropes, implantation of a ventricular assist device or even
cardiac transplantation may become necessary. The role of immunosuppressive
therapy has not borne out and remains limited to myocarditis clearly
attributable to systemic autoimmune disease. Antiviral agents such as interferon
beta are under evaluation at this time and have shown initial promise.
Infective
Endocarditis
In the last thirty years, the etiology of infective endocarditis has shifted
with the eradication of rheumatic fever in much of the industrialized world.
While valvular diseases such as mitral valve prolapse and mitral regurgitation
remain key risk factors for infective endocarditis, skin flora, primarily
staphylococci, now surpass oral streptococci (viridans group streptococci) as
the leading cause of infective endocarditis. In particular, Staphylococcus aureus has emerged as the predominant organism responsible
for most new cases of infective endocarditis worldwide, with a significant share
attributable to methicillin-resistant S. aureus. Together with Enterococcus spp., staphylococci and streptococci comprise more than 80% of
all causes of infective endocarditis. Intravenous drug users may also be at
heightened risk for infection with Pseudomonas aeruginosa and fungi. In
addition to the typical organisms, elderly persons with degenerative valvular
disease may be more prone to infection with Streptococcus bovis, which
has been associated with gastrointestinal malignancy. Patients with prosthetic
valves may develop infective endocarditis from coagulase-negative staphylococci
and gram-negative bacteria of the HACEK group. Patients with nosocomial or
healthcare-related infections from invasive surgical procedures, infected
hardware and long-term hemodialysis represent a growing population at risk for
staphylococcal and enterococcal endocarditis as well.
Fever remains the most common clinical presentation of infective endocarditis.
While the fever may be remitting in nature, it may exceed 40º C in cases of
acute infective endocarditis and can be accompanied by chills and rigor. It may
also be absent in the elderly and in patients with congestive heart failure,
chronic renal failure, liver disease or infective endocarditis caused by less
virulent organisms. Systemic symptoms such as myalgias, fatigue, malaise, night
sweats, anorexia, nausea and vomiting may be seen and often predominate in
subacute presentations. Up to 10% of patients with infective endocarditis may
report chest pain. Heart murmurs may be heard in as much as 85% of patients. In
many instances, these murmurs are preexisting. However, a new, altered or
changing murmur has a strong predictive value for infective endocarditis when
taken in context with established bacteremia. Peripheral embolic manifestations
can be nonspecific for infective endocarditis and are increasingly infrequent
due to earlier diagnosis and treatment. Classic Janeway lesions are nontender,
erythematous vesicles or pustules seen on the palms and soles while Osler’s
nodes are painful, subcutaneous nodules found on the pulp of the digits.
Splinter hemorrhages may be seen in the nail beds of the fingers and toes.
Petechiae may be seen in the conjuctiva and buccal mucosa. Pale, oval lesions
surrounded by hemorrhage, known as Roth’s spots, may be noted on examination of
the retina. Splenomegaly may be evident in delayed presentation or late
diagnosis of endocarditis.
Blood cultures remain the cornerstone of diagnosis for infective endocarditis.
Three or more sets of blood cultures should be drawn at least one hour apart
(fresh stick each time preferable) prior to initiation of antibiotics. Cultures
may be drawn regardless of whether the patient is febrile. Blood cultures may be
negative in about 15% of cases of infective endocarditis. While this may related
to prior administration of antibiotics, fastidious organisms such as Coxiella
burnetti (Q fever), HACEK organisms, and fungi may require additional
serologic or molecular techniques to identify. Laboratory evaluation is
otherwise nonspecific. Inflammatory markers such as the erythrocyte
sedimentation rate and C-reactive protein may be elevated. Leukocytosis and a
normocytic anemia may or may not be present, depending on the severity of the
presentation. Urinalysis may reveal proteinuria, hematuria or casts suggestive
of an immune complex glomerulonephritis that may be seen with infective
endocarditis.
All patients with suspected infected endocarditis should have an
electrocardiogram performed. New atrio-ventricular, fascicular or bundle-branch
block may signify perivalvular invasion and potential abscess formation. Aortic
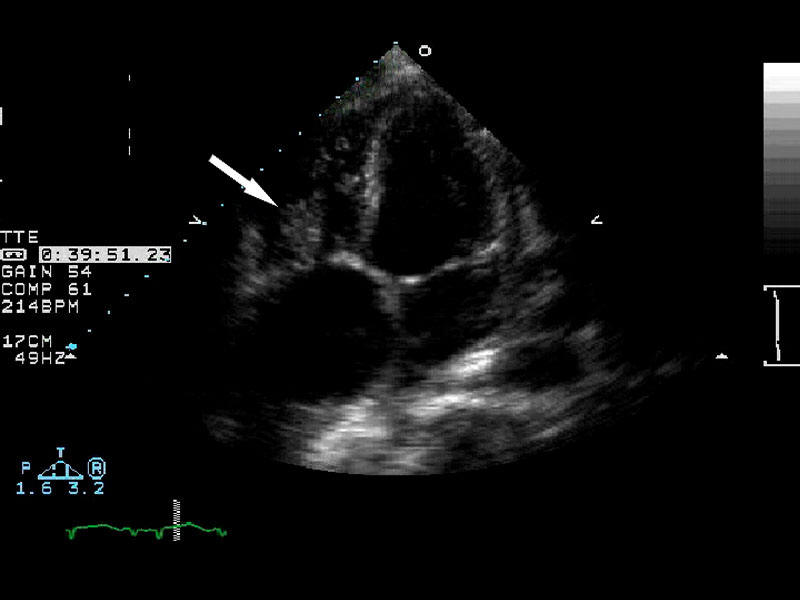
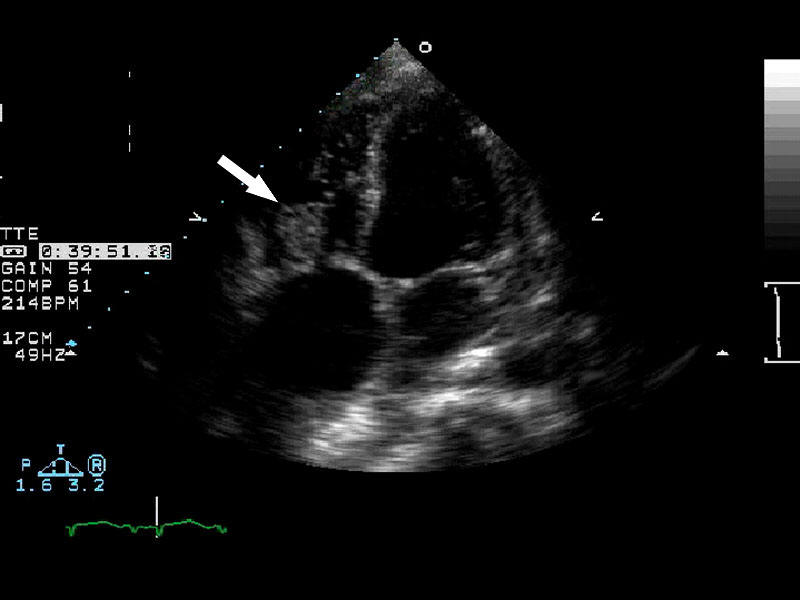
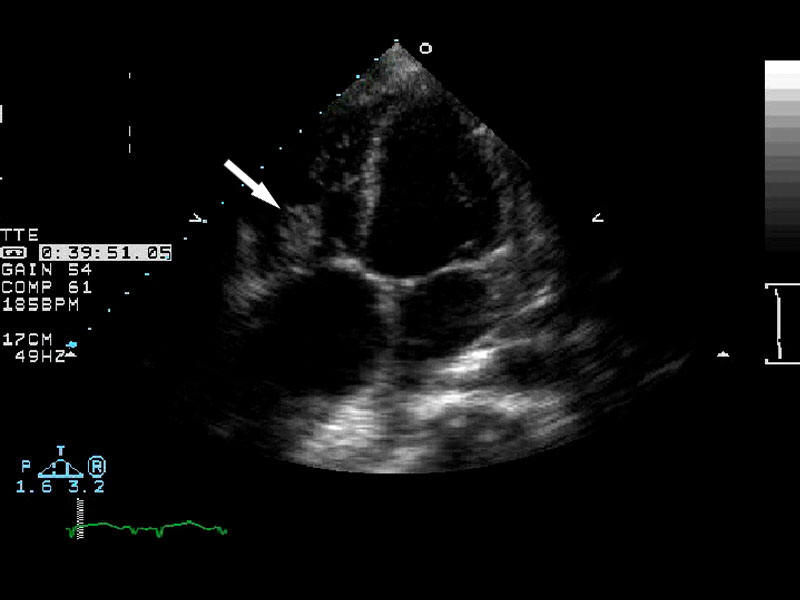
valve involvement is most common. Echocardiography should be employed to
evaluate for intracardiac masses or vegetations
, valve competence and myocardial
abscess. It is reasonable to perform a transthoracic echocardiogram (TTE) first
if the patient is clinically stable or if there is low clinical suspicion for
infective endocarditis. Transesophageal echocardiography (TEE) should be
considered if the patient is a difficult imaging candidate for TTE or there is
moderate to high clinic suspicion for infective endocarditis. A negative TTE in
the setting of deteriorating clinical course should prompt TEE in light of its
higher sensitivity for identifying vegetations and abscesses. False-negative
results in both TTE and TEE may occur if vegetations are small or have embolized.
Large, mobile vegetations (> 10 mm), particularly on the anterior mitral
leaflet, have the greatest potential to embolize and have been linked with
increased mortality.
|
Arrow
demonstrating vegetation on the heart valve diagnostic of endocarditis. |
|
 |
 |
 |
Remarkably, the four elements characterizing infective endocarditis first
described by Sir William Osler in 1885 remain relatively unchanged: persistent
bacteremia with an appropriate infectious microorganism, predisposing factors,
active endomyocardial involvement and vascular phenomena. Given the protean
nature of its presentation, multiple criteria have evolved throughout the years
to aid with accurate diagnosis. The widely accepted Duke criteria provide a
framework for diagnosis of infective endocarditis that has a sensitivity of
roughly 80% (see Table 3).
In 2005, the American Heart Association published its most recent guidelines
regarding antibiotic therapy for infective endocarditis, which has since been
endorsed by the Infectious Disease Society of America (IDSA). Therapeutic
recommendations are available in the endocarditis chapter of Empiric.
Table 3: Modified Duke Criteria for
the Diagnosis of Infective Endocarditis
|
Major criteria
Blood culture positive for IE
Typical microorganisms for IE
isolated from 2 separate blood cultures (Staphylococcus
aureus, Viridans streptococci, Streptococcus bovis, HACEK group, or community-acquired enterococci without
a primary focus)
or
Micoorganisms consistent with
IE from persistently positive blood cultures
or
Single positive blood culture
for Coxiella burnetti or phase I IgG antibody titer >1:800
Evidence of endocardial
involvement
Echocardiogram positive for
IE (oscillating intracardiac mass on a valve or supporting structures, paravalvular abscess or new dehiscence of a prosthetic valve)
or
New valvular regurgitation
(worsening or change in pre-existing murmur is not sufficient) |
|
Minor criteria
Predisposing factors for IE
(e.g. valvular disease, injection drug use) Fever > 38º C
Vascular phenomena (major
arterial emboli, septic pulmonary emboli, mycotic aneurysm,
intracranial hemorrhage,
conjunctival hemorrhages, and Janeway lesions)
Immunologic phenomena
(glomerulonephritis, Osler’s nodes, Roth’s spots and rheumatoid factor)
Microbiological evidence
(positive blood culture not meeting major criteria or serologic
evidence of active
infection with an organism consistent with IE) |
|
Definite infective endocarditis
2 major criteria
1 major criteria and 3 minor
criteria
5 minor criteria |
|
Possible infective endocarditis
1 major criteria and 1 minor criteria
3 minor criteria
|