
Patients with Recurrent Infections
Authors: David R. Stukus, M.D., Mark E. Rose, M.D., David M. Lang, M.D.
The purpose of this chapter is to present an overview of the clinical presentation, diagnosis, and management of a patient who presents with recurrent infections. For a more comprehensive review or details regarding the known primary immune deficiencies, please visit the references and/or online resources listed at the end of this chapter.
Definition of Recurrent Infections
It is difficult to assign a precise frequency of infections that defines an increased susceptibility to infections that reflects an impaired immune response. For example, the majority of patients who have intact immune systems may still contract multiple upper respiratory infections each year, usually of viral origin. The frequency of these infections may be related to exposures, as in health care and daycare workers, teachers, and parents, who are routinely exposed to children or other individuals who may transmit viral infections, particularly during the winter months. The average child may experience up to six to eight upper respiratory infections each year, most commonly of viral origin (34). In addition, individuals who are under high stress, have poor sleep hygiene, tobacco smoke exposure, co-morbid conditions such as diabetes, or need to take chronic immune modulating medications (e.g., oral corticosteroids) may experience immune system dysregulation and a predisposition for infection (6,23,14). Many patients are treated for suspected bacterial infection, especially of the upper or lower respiratory tract, but may actually have allergic rhinitis or self limited viral infections. Without objective data obtained via imaging studies, biopsy, or gram stain and culture of relevant secretions to confirm a diagnosis, it is not possible to know how many patients truly have recurrent bacterial infections as opposed to common viral upper or lower respiratory infections. However, it may not be feasible from the standpoint of the patient and/or physician to obtain such evidence. For instance, antral tap through the maxillary sinus is considered the gold standard for obtaining a sinus culture to direct antimicrobial treatment of sinusitis, but this is commonly not performed. For this reason, patients with (suspected) sinusitis are commonly treated based upon their presentation, clinical suspicion, and knowledge of bacterial organisms most frequently implicated as causes of sinusitis. Even with appropriate imaging studies, it may still be difficult to reliably assess these findings -- such as when the clinician is faced with distinguishing bacterial pneumonia from atelectasis with fever in a child with reactive airways disease. Not infrequently the clinician may find it difficult to distinguish patients with an intact immune response and normal pattern of infections (or non-infectious etiologies) from those with a presentation reflecting primary or secondary immunodeficiency. In addition to considering the absolute number of infections during a period of time, such as one year or several years, there are other clues which also may signify an impaired immune response. These are detailed in Table 1 (44).
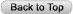
Epidemiology
Primary immune deficiencies are rare in the general population, with estimates of overall incidence ranging from 1 in 2,000 to 1 in 10,000 live births, with a prevalence of 1 in 10,000 in the general population (53,61). Primary immunodeficiency results from genetic defects which may arise through various inheritance patterns or may be the result of spontaneous mutation. The true incidence of these disorders is unknown, as no prospective study has yet to be conducted. Estimated incidence rates vary from the most common inherited immunodeficiency, Selective IgA Deficiency (1 in 300 to 700 live births), to very rare disorders such as Chronic Granulomatous Disease (1 in 200,000 live births) (5). The prevalence of secondary immunodeficiency, e.g., immune dysfunction due to drugs or infection (HIV), is also unknown.
Due to the rarity of immune deficiency in the general population, the negative predictive value of laboratory tests is more optimal than positive predictive value (44). Thus, it is important to carry out diagnostic evaluation of immune function in patients with a history of recurrent, invasive, or opportunistic infections who have a higher pretest probability of impairment in immune function. Otherwise, a practitioner may be faced with abnormal values that may have no or uncertain clinical significance.
Pathogenesis
Immunodeficiencies are classified according to the principal immunologic mechanisms that are abnormal and fall into four main classes (Table 2):
1. Humoral or antibody immunodeficiency is the most common form, accounting for approximately half of all primary immunodeficiency (52, 53, 61). Humoral immunity, which is part of the adaptive immune response, is largely responsible for clearing bacterial infections. It is comprised of the five types of immunoglobulin, listed here in order of decreasing serum concentrations, IgG, IgA, IgM, IgE, and IgD. Patients with primary humoral immune deficiency may present at any age, often not until the 3rd, 4th, or even 5th decade of life (59).
2. Cellular immunodeficiency results from dysfunction of T cells, which are principally involved in host defense for viral, mycobacterial, and fungal infections. T cells also play a major role in the adaptive immune response and are intimately involved in the recruitment and activation of B cells, providing stimulation of antibody production (57). Some types of humoral immunodeficiency actually result from a defect in T cell signaling, which prevents separate antibody isotypes from forming.
Combined B and T cell deficiency, such as Severe Combined Immunodeficiency, is very rare and often presents in infancy with chronic diarrhea, failure to thrive, and severe, recurrent, opportunistic infections in the first few months of life. These patients require a bone marrow or stem cell transplant in order to survive, as this immunodeficiency state is often fatal in the first year of life if left untreated.
3. Phagocytic disorders result from ineffective recruitment or function of neutrophils, phagocytes, and natural killer cells, which are all part of the innate immune response. Phagocytes are responsible for engulfing and destroying bacterial organisms through recognition of certain cell wall constituents. These disorders are rare, comprising roughly 10-20% of all primary immunodeficiency.
4. Complement deficiency is the rarest form of immunodeficiency, comprising less than 1% of all cases (5). The complement system is comprised of nine principal proteins, C1-9, as well as a number of regulatory proteins, and is activated by three separate mechanisms, the classical, lectin, and alternative pathways (58). This system was aptly named as these proteins ‘complement’ the antigen binding function of antibodies in host defense against bacterial pathogens.
Clinical Manifestations
Patients with immune deficiency characteristically present with recurrent infections of varying severity and anatomic location (Table 3). However, the same immunologic defect or dysregulation that predisposes the patient to infection may concomitantly lead to autoimmune or inflammatory disease (27). Vasculitic disorders, autoimmune cytopenias, and inflammatory arthropathies are a few examples of autoimmune conditions commonly associated with various types of immune deficiency (11,33,56). In addition, malignancies, such as leukemia and lymphoma, occur with increased frequency in association with common variable immune deficiency (12). Infectious pathogens associated with primary immunodeficiency are listed in Table 4.
Patients with humoral immune deficiency present with repeated pyogenic infections with gram positive encapsulated bacteria such as Streptococcus pneumoniae, Haemophilus influenzae, and Staphylococcus aureus as well as gram negative organisms such as Pseudomonas (41). They do not typically succumb to opportunistic, viral, or fungal infections but may have increased susceptibility to enteroviral infections, specifically meningoencephalitis. The anatomic location most frequently affected is the sinopulmonary tract, and these patients have frequent/recurrent episodes of sinusitis, otitis media, and pneumonia.
T cell deficiencies or dysfunction may predispose patients to recurrent or severe viral, mycobacterial, or fungal infections (57). Hyper IgM syndrome, which is due to a T cell deficiency, is characterized by an impaired humoral immune response due to signaling errors and inefficient antibody recruitment or activation.
Phagocytic disorders are rare and may have variable presentations. Chronic Granulomatous Disease (CGD) results from a defect in the oxidative burst used by phagocytes in killing engulfed bacteria. These patients are susceptible to infections with catalase producing organisms such as Staphylococcus aureus and Klebsiella. There is often a history of recurrent skin abscesses, lymphadenitis, or necrotizing pneumonia. These patients may also have a history of gastrointestinal or bladder disturbance due to the formation of granulomas along the intestinal or genitourinary tract. Leukocyte Adhesion Deficiency results from a defect in an adhesion protein utilized in the recruitment of phagocytes during times of inflammation or infection. These patients may have extremely elevated peripheral white blood cell counts (above 100,000) due to the inability of the white blood cells to extravasate through the vascular endothelium to sites of infection. Patients with LAD cannot produce pus at the site of infection and may have a history of delayed separation of the umbilical cord (longer than 4-6 weeks).
Disorders of the complement cascade may or may not result in increased susceptibility to infections. The most common presentation of complement deficiency is autoimmune disease, especially Systemic Lupus Erythematosis (42). However, deficiency of the early complement proteins (C1, C4, C2) may lead to increased susceptibility to pyogenic infections with encapsulated bacteria, whereas deficiency of terminal complement proteins (C5-C9, or the Membrane Attack Complex) may lead to recurrent infections with Neisseria sp (19). C1 inhibitor deficiency predisposes affected individuals to experience episodic angioedema of the deep dermal tissues in the GI tract, airway, extremities, or facial areas. This can be hereditary (autosomal dominant) or acquired. The latter condition is quite rare, and reflects presence or may be the harbinger of an underlying neoplastic or autoimmune disease (13).
Differential Diagnosis
The evaluation of a patient with recurrent infections must include a careful assessment for the presence of other acute or chronic illnesses. Secondary immune deficiency or dysfunction may not improve unless an underlying condition is appropriately identified and treated. As mentioned previously, primary immune deficiency is rare in the general population, therefore a thorough evaluation of other possible causes is essential.
Allergic inflammation of the upper respiratory tract may predispose patients to frequent bacterial infections such as otitis media and sinusitis. In addition, patients with chronic rhinitis and/or asthma, including those with aspirin exacerbated respiratory disease, may have elevated rates of chronic sinusitis compared with the general population (43,26).
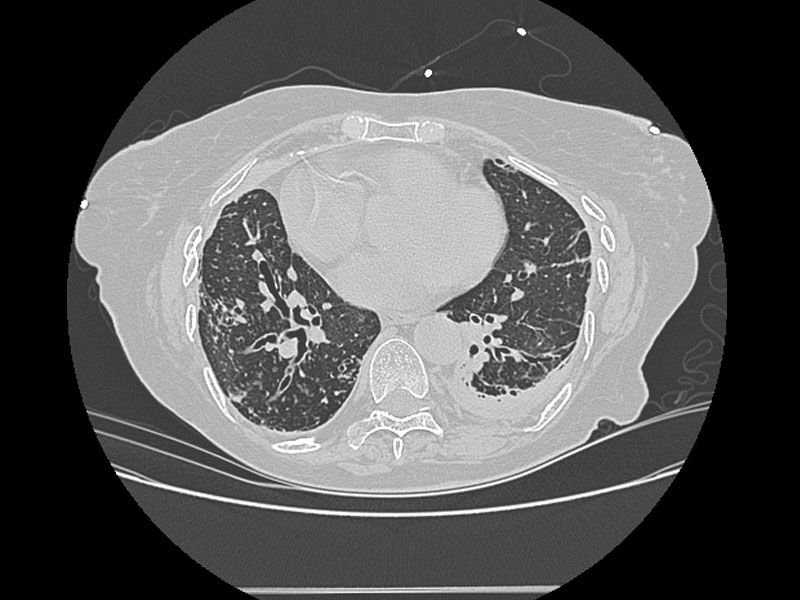
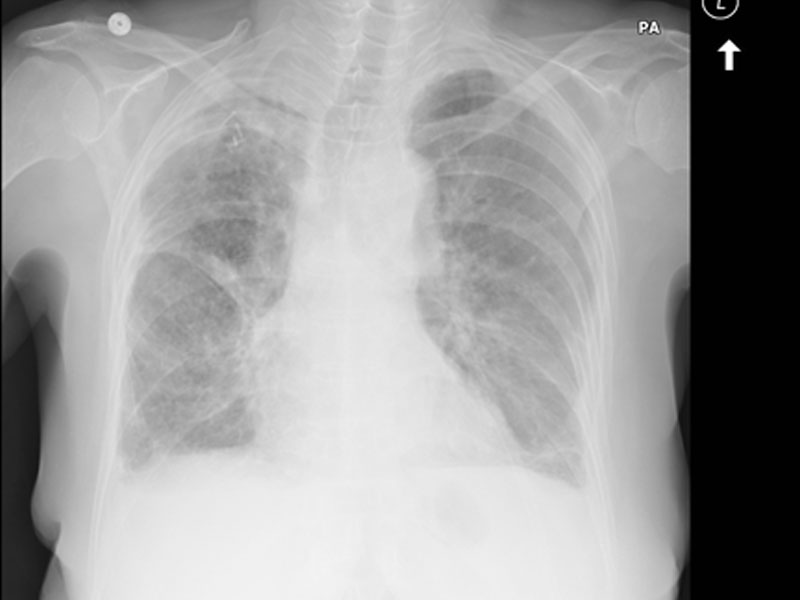
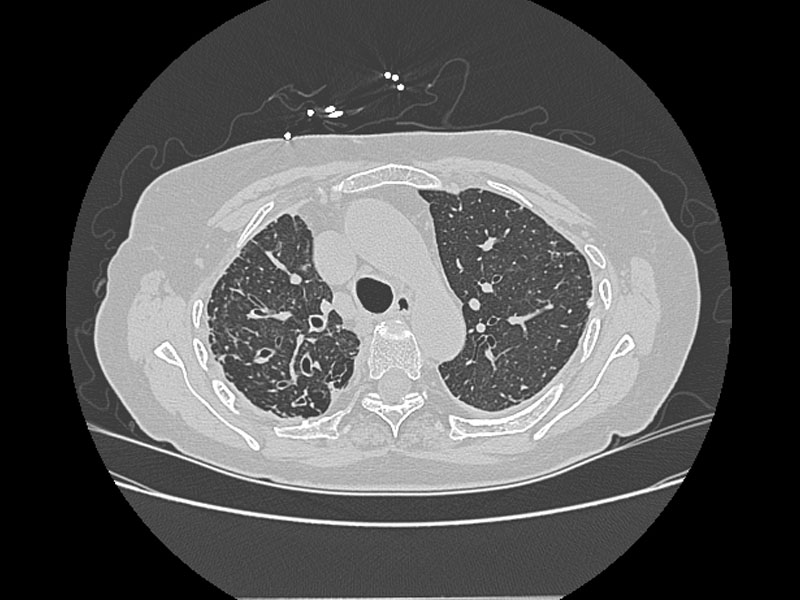
Alterations in lung anatomy, as seen in COPD with bronchiectasis 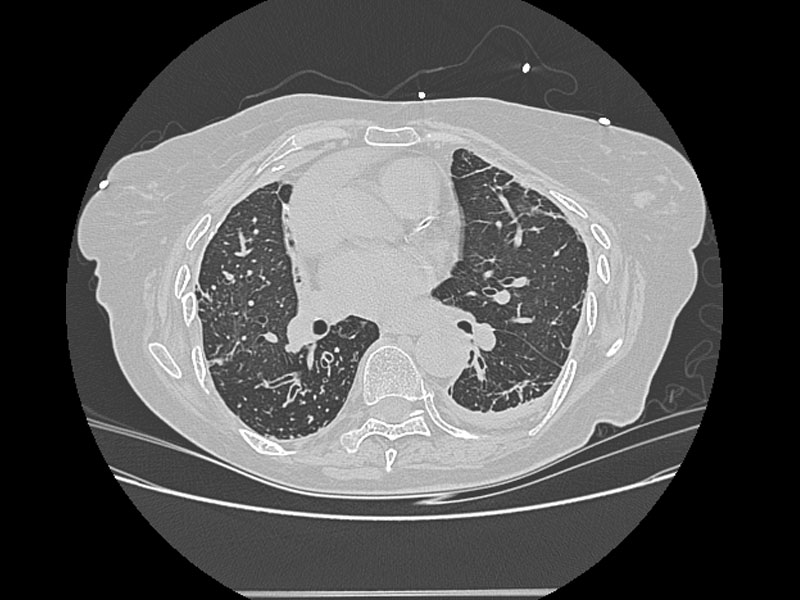 , may predispose patients to recurrent pneumonia and lower respiratory tract infections. Patients with cystic fibrosis and ciliary dyskinesia are also susceptible to recurrent respiratory infections (32).
, may predispose patients to recurrent pneumonia and lower respiratory tract infections. Patients with cystic fibrosis and ciliary dyskinesia are also susceptible to recurrent respiratory infections (32).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diabetes mellitus can lead to neutrophil dysfunction and predisposition to fungal and bacterial infections (14). Poor peripheral circulation in diabetic patients may lead to skin ulceration and diminished delivery of neutrophils to sites of microbial entry.
Immunoglobulin loss from protein losing enteropathies and nephrotic syndrome can predispose patients to more frequent bacterial infections (21,37). Patients undergoing dialysis may display reduced T cell function, diminished antibody production, and poor neutrophil function; requirement for an indwelling catheter offers a nidus for infection, and may also result in recurrent infection unrelated to a primary immunodefiency state (14,28).
Cirrhosis may predispose patients to sepsis and bacterial peritonitis from complement deficiency and dysfunction (14).
Malnutrition secondary to any co-morbid conditions may lead to impaired specific antibody and T cell response and phagocyte dysfunction (45).
Asplenia or hyposplenism from any cause is associated with increased risk of infection with polysaccharide encapsulated bacteria (16).
Autoimmune disease can increase susceptibility to infections, especially in systemic lupus erythematosus (40,24). Trauma and burns may lead to immune dysfunction secondary to a massive release of inflammatory cytokines and widespread activation of monocytes and macrophages (17,3).
Resistant organisms such as Methicillin-Resistant Staphylococcus aureus (MRSA) have become more prevalent in recent years. Patients may present with recurrent skin abscesses, furuncles, or boils which may be related to close contact with a family member or caretaker colonized with MRSA. These infections tend to recur and respond poorly to standard antimicrobial agents. In addition, any patient who has recently received care as an inpatient in a hospital environment may also be susceptible to nosocomial infections such as Vancomycin Resistant Enterococcus (VRE).
A number of malignancies, including chronic lymphocytic leukemia, lymphoma, and multiple myeloma are associated with secondary hypogammaglobulinemia (1,9,39).
HIV infection must also be considered in any patient with severe recurrent or opportunistic infections.
The process of aging may not only lead to increased incidence of acute and chronic illness, but increased use of medications and deteriorating nutritional status. Impaired innate barrier function in the elderly also occurs with decreased mucous secretion and loss of tissue elasticity and/or contractility. Phagocytic cell function may decrease and while the number of circulating immunoglobulins may remain constant, specific antibody titers may fall as autoantibody formation rises (46). Elderly patients may also have more inconsistent antibody response to vaccination, with a more rapid waning of response, compared with younger patients (2).
Many patients may not have overt signs or symptoms of these or other underlying illnesses and yet may still present with recurrent infections. Other factors to consider include medications, physical or emotional stress, history of tobacco use, and illicit drug use. Several medications can cause reversible secondary hypogammaglobulinemia, including sulfasalazine (18), gold (50), corticosteroids, phenytoin (54), carbamazepine (35), and androgen replacement therapy (60). Major life stresses such as bereavement, and even less catastrophic stresses such as medical school examinations, have been associated with increased rates of respiratory tract infection, reactivation of herpes virus infections, and increased incidence of cancer (25,22). Tobacco smoking may lead to chronic inflammatory changes in the mucosa of the upper and lower respiratory tract as well as ciliary dysfunction. Illicit drug use may result in not only malnutrition, but also increased physical and emotional stress.
Laboratory Evaluation
Any patient with suspected immunodeficiency should have baseline laboratory tests such as a complete blood count with differential and complete metabolic panel. Then, depending upon the history, types of infections, and pretest probability, the four separate arms of the immune system can be formally evaluated, as described below. As mentioned previously, the positive predictive value of these tests is lower than the negative predictive value, and for this reason, not every patient will require, nor should have, a full evaluation of all four arms of the immune system. Abnormal results must be interpreted with caution and in the context of clinical presentation.
Humoral Immune System
Serum immunoglobulin IgG (including subclasses IgG 1, 2, 3, 4), IgA, and IgM concentrations are measured using rate nephelometry or with an enzyme linked immunosorbent assay, which are sensitive, reliable tests that quantify antibody levels (44). Levels are reported with normal values in units of milligrams per deciliter and values that are more than 2 standard deviations from the mean are considered abnormal (Table 5).
Humoral immune function should be assessed by measuring antibodies to protein and polysaccharide antigen before and after immunization (36,20). First, blood should be obtained for antibodies to protein and polysaccharide antigens such as tetanus and pneumococcus. If the initial titers are low, then the patient should be re-vaccinated, even if they recently received the vaccine as many patients, particularly young children or elderly, may have poor natural response (30). Four weeks after vaccination, blood should be re-tested for antibody titers. There is a range of antigens to which normal individuals will respond and a four-fold rise in specific antibody titers 4 weeks after vaccination is consistent with an intact humoral immune response (5). No controlled study has determined the number of antigens to which one should respond, but a lack of response to all antigens is consistent with impairment of the humoral immune system. The recently developed pneumococcal conjugate protein vaccine (Prevnar) has complicated evaluation of humoral immune response. Its use for testing is controversial and is not generally recommended, as antibodies may develop to the conjugated protein but not to capsular antigens (44).
Cellular Immune System
Lymphocyte enumeration studies are useful for quantitation of CD4, CD8, and NK cells. The levels of these T cells are typically reported with a range of normal values that may differ depending upon which specialty laboratory is performing the analysis. Levels that fall more than 2 standard deviations outside the mean are reported as abnormal. These values must be interpreted with caution and in the appropriate clinical context, as abnormal values for specific T cell subpopulations may be seen in lymphopenic states or may result from improper specimen collection and/or handling. Steroids and stress can lead to false abnormalities in T cell numbers and should be performed when patients are not acutely ill and repeated to verify abnormal results.
Lymphocyte function may be studied by either in vitro or in vivo methods. In vitro lymphocyte responses to mitogens are nonspecific and indicate the ability of T cells to be simulated by powerful stimuli (5). Normal ranges for appropriate T cell response to mitogens are usually specific to the laboratory that is performing the study. A useful in vivo method to evaluate T cell response to specific antigen is an anergy panel, or cutaneous delayed hypersensitivity response (20). This test is similar to the purified protein derivative reaction used for tuberculosis evaluation but utilizes recall antigens such as tetanus, candida, or mumps. These antigens are injected intracutaneously and a normal response would entail erythema and at least 2 to 5 mm of induration 48 to 72 hours after placement (5). A diminished response to cutaneous delayed hypersensitivity testing does not necessarily indicate an impaired cell mediated response as this can be seen in very young patients and in patients receiving chronic immunosuppressive therapy (e.g., oral corticosteroids).
Phagocytic Evaluation
As discussed previously, phagocytic disorders are extremely rare and these specialized tests should be ordered and interpreted by a physician who has experience in the evaluation and management of patients with primary immune deficiencies. The traditional screening test for Chronic Granulomatous Disease (CGD) is the Nitroblue tetrazolium (NBT) test. This assesses presence and function of the NADPH oxidase system, which is impaired in neutrophils of patients with CGD. The assay verifies the ability of cells to convert NBT, which is a colorless chemical, to a deep blue color. The sample collection method and technique used for the NBT test is very sensitive and may result in false positive results.
In recent years, genetic mapping has identified several loci responsible for these rare diseases, including CGD and Leukocyte Adhesion Deficiency. Various molecular diagnostic tests are now available through individual laboratories that specialize in primary immune deficiencies which may contribute to the diagnosis of such defects (see links below for online resources).
Complement System
The CH50 is a validated in vitro screening test for overall function of the complement system (58). This is a functional assay of total complement activity that measures the capacity of serial dilutions of serum to lyse a standard preparation of sheep red blood cells coated with anti-sheep erythrocyte antibody. The reciprocal of the dilution of serum that lyses 50 percent of the erythrocytes is reported as the whole complement titer in CH50 units per milliliter of serum. A result that falls within the normal range is consistent with an intact classical complement cascade. However, abnormal results cannot accurately identify which complement protein(s) may be deficient or dysfunctional. Further evaluation with measurement of individual complement protein levels may be necessary. In addition, the initial factors involved in the alternate complement cascade are not measured with a CH50 assay but can be more accurately assessed by using a similar AH50 assay.
Whenever possible, it is appropriate to identify the precise genetic defect in any patient with primary immune deficiency to provide unequivocal diagnosis, genetic counseling, future pregnancy planning, and to target individuals who are candidates for gene specific therapies (5).
Treatment
Early diagnosis and treatment of patients with immune deficiency are important to prolong survival, improve quality of life, and prevent significant end organ damage or death from infection. Immunodeficient patients often require more aggressive treatment of infections and may not respond to standard dosing or duration of antimicrobial agents. Whenever possible, culture data should be sought to direct proper antimicrobial treatment options. Many patients with recurrent infections may also benefit from the use of daily or alternate day prophylactic antibiotics. Once a primary immune deficiency has been specifically identified and confirmed, then additional targeted therapies may be available and indicated depending upon the defect that is present.
Patients with impaired antibody production, such as Common variable immunodeficiency (CVID), X-linked agammaglobulinemia, or Severe combined immunodeficiency (SCID), should receive immunoglobulin replacement therapy (5,38,51). The utility of intravenous immune globulin (IVIG) for treatment of recurrent infections has only been established for this subset of patients. IVIG has no established benefit for the treatment of IgG subclass deficiency, selective IgA deficiency, COPD with hypogammaglobulinemia but normal antibody function, or patients with hypogammaglobulinemia and no history of recurrent infections (which may represent a normal subset of the general population, especially in children) (Table 6) (,5,38,44,51). Immune globulin is pooled from the sera of thousands of screened donors and multiple different products are available. Products vary in osmolality, concentration of IgG and IgA, and cost. Treatment with immune globulin replacement will not improve specific antibody response to protein or polysaccharide antigens. Any patient with an absolute IgA deficiency may form IgE antibodies directed at IgA and must receive an immune globulin product that is IgA-depleted, to avoid risk of sensitization and subsequent anaphylaxis (44). There is a risk of anaphylaxis for any patient receiving IVIG, which is greatest with the first few infusions. Infusions are typically given every 3-4 weeks and require intravenous access for IVIG. The establishment of permanent central venous access for the sole purpose of administration of IVIG is generally discouraged (7).
Patients with impaired antibody production who receive regular immune globulin replacement therapy will not be cured of their underlying immune deficiency, will often continue to suffer from recurrent infections, and may still require frequent or prophylactic antibiotics (8, 41). However, evidence indicates the frequency and severity of infections is typically reduced and quality of life and length of survival are improved with regular administration of immune globulin (8).
In addition to anaphylaxis, there are other potential risks associated with the administration of immune globulin. Immune globulin is a blood product and although these products undergo screening protocols similar to other blood bank products, there is risk of viral contamination (38,44,51). Many patients will experience side effects during or after the infusion, particularly with the first few infusions or with a change to any new immune globulin product (38,51). These can often be controlled by slowing or stopping the infusion and with treatment and/or premedication with non steroidal anti-inflammatory drugs and antihistamines (38,51). Commonly reported symptoms include chills, rigors, hypotension, lightheadedness, myalgias, low grade fever, headache, and occasionally shortness of breath and/or dyspnea (5, 38,44,51). Patients may also experience delayed adverse reactions 24 to 72 hours after the infusion, including aseptic meningitis (47). After the diagnosis of primary immune deficiency with impaired antibody production has been established, then therapy with immune globulin replacement is life long unless a definitive treatment is available, e.g. stem cell transplantation in patients with SCID.
Any patient who is receiving regular immune globulin replacement and does not meet specific criteria as outlined above, should do so on a trial basis. Well defined end points must be established prior to starting treatment and may include a reduction in frequency or severity of infections (with careful documentation of any suspected bacterial infections, including appropriate imaging and/or culture data), number of hospitalizations due to infection, reduced number and/or duration of antibiotic courses, or improved quality of life (5). Patients should be followed closely and therapy should be discontinued if these end points are not reached within 9-12 months.
The definitive treatment of cell mediated and combined B and T-cell immune deficiencies requires hematopoietic stem cell transplantation (5,7). The best outcomes are typically achieved by using HLA-matched sibling donors, which is not always possible. In addition, treatment before the age of 3 months is also associated with better prognosis. Any patient with suspected or confirmed T-cell or combined immune deficiency should be referred to a tertiary care facility that has experience in hematopoietic stem cell transplantation and the treatment of primary immune deficiencies (5).
Patients with cellular immune deficiency who require blood products should receive irradiated, CMV-negative, lymphocyte depleted products (5). In addition, live vaccines are absolutely contraindicated in these patients as disseminated disease has been observed in severely immunocompromised patients who have received these vaccines (5,48). This currently includes measles-mumps-rubella, varicella, yellow fever, oral polio virus, oral typhoid, andBCG vaccines.
Patients with phagocytic or complement deficiency must be monitored closely for any signs or symptoms of infection. As with other immune deficiencies, antibiotics are often used as treatment or prophylaxis for recurrent infections. Depending upon the specific genetic defect, other therapies may be available, e.g. interferon gamma with CGD (7). There is currently no role for hematopoietic stem cell transplantation or immune globulin replacement in the treatment of these patients.
Indications for Antibiotic Prophylaxis
There are no randomized, controlled trials to date that have evaluated the efficacy of using prophylactic antibiotics in patients with known or suspected primary immune deficiencies. Studies conducted with neutropenic patients, mostly patients with leukemia undergoing induction chemotherapy or status-post bone marrow transplantation, have yielded conflicting results regarding the efficacy of prophylactic antibiotics (55, 56). The CDC has made recommendations for prophylaxis against Pneumocystis Carinii pneumonia with one double-strength Bactrim tablet daily in HIV patients who have absolute CD4 counts less than 200 cells/ul(10). These recommendations may be extrapolated to other patients with a similar deficiency in CD4+ cells, such as in SCID, but there are no studies in support of this. For a patient with recurrent infections due to one particular type of microorganism, e.g. recurrent sinusitis due to Streptococcus pneumoniae, culture directed antimicrobial prophylaxis may be warranted. Unfortunately, there are no well established guidelines regarding what antibiotic class or dosage is most appropriate in this setting. Increased rates of antimicrobial resistance, risk for adverse effects, and cost of prolonged use of antibiotics must all be considered. For these reasons, routine antimicrobial prophylaxis is not generally recommended for a patient who presents with recurrent infections and should be reserved for patients who have been thoroughly evaluated for possible immune deficiency and who have also failed conventional medical management.
Prognosis
Any patient with suspected or documented immune deficiency warrants evaluation and management with a physician who has experience in managing such conditions. Patients with borderline hypogammaglobulinemia but no evidence of primary immune deficiency may need to have immunoglobulin levels followed routinely every 6-12 months, with careful surveillance and treatment of infections. Vaccinations (e.g., influenza and pneumovax) should be administered as indicated. Any patient who is receiving IVIG should have serum immunoglobulins monitored every 6-12 months and have the dose and/or frequency of infusions adjusted to maintain total serum IgG levels ≥ 500 mg/dL.
Patients with immune deficiencies may continue to experience infections, although at reduced frequency, despite appropriate management -- including intravenous immunoglobulin (8, 41). Long term morbidity and end organ damage relating to repeated episodes of infection can occur (5), e.g. development of permanent lung parenchymal damage with bronchiectasis in patients who experience frequent episodes of pneumonia. A number of primary immune deficiencies are associated with an increased rate of autoimmune and malignant disorders (12,16,27,33,56). Regular follow up and screening for these conditions is imperative in any patient with primary immune deficiency.
Long term survival is decreased in patients with primary immune deficiency. In one large series of patients with CVID, survival 20 years after diagnosis was 64 and 67 percent for males and females, respectively, compared to an expected 92 percent population survival for males and 94 percent for females (11). Long term survival data are not available for many of the more rare cellular, combined, phagocytic, and complement disorders, but these patients typically suffer increased morbidity due to complications of either the disease process itself or various treatment modalities, including hematopoietic stem cell transplantation.
Summary
Patients who present with recurrent infections warrant careful evaluation regarding the type of infection, frequency, anatomic location, and response to standard treatment modalities. Primary immune deficiencies are rare in the general population, and the majority of patients with recurrent infection will not have evidence of impaired immune system response. Recurrent infections of unusual severity or anatomic location merit further evaluation. A careful search for other factors that may predispose to infection, such as barrier abnormalities, allergic rhinitis, and other co-morbid conditions, is warranted. Laboratory studies, including a complete blood count with differential, complete metabolic profile, and other indicated screening tests, should be obtained. Depending upon the types of infection (bacterial, viral, fungal), and index of suspicion, a formal evaluation of the functional capacity of the patient’s immune system may be indicated. Therapy with prophylactic antibiotics or, in patients with impaired antibody production, immunoglobulin replacement with IVIG may be beneficial in properly selected patients. Prognosis may also depend upon accurately identifying and treating underlying conditions that may increase susceptibility to infection and influence response to treatment.
Resources
Several online resources are available for more information regarding primary immune deficiencies:
The Immune Deficiency Foundation (IDF) and Jeffrey Modell Foundation (JMF) are excellent resources for both physicians and patients/families. The IDF has a program available where “Counseling Immunologists” are available for second opinions by reaching (877) 666-0866 during weekdays. The IDF website also has links to the Practice Parameter for the Diagnosis and Management of Primary Immune Deficiency. The “10 Warning Signs of Primary Immunodeficiency” poster is available through the JDF website.
The following website offers information regarding laboratories, both clinical and research that offer genetic testing and/or mutation analysis for different immune deficiencies: Gene Tests.
The American Academy of Asthma, Allergy, and Immunology also provides excellent resources on its website, including a PDF download of the Practice Parameter for the Diagnosis and Management of Primary Immune Deficiency.
References
1. Aittoniemi J, Miettinen A, Laine S, et al. Opsonising immunoglobulins and mannan-binding lectin in chronic lymphocytic leukemia. Leuk Lymphoma 1999; 34:381-385. [PubMed]
2. Armitage KB, Duffy EG, Mincek MA, et al. Transient normalization of lymphocyte blastogenic and specific antibody responses following boosting of healthy elderly subjects with tetanus toxoid. J Gerontol 1993; 48:M19.[PubMed]
3. Barlow Y. T lymphocytes and immunosuppression in the burned patient: a review. Burns 1994; 20:487.[PubMed]
4. Bartlett J. Breiman R. Mandell L. File T. Community-Acquired Pneumonia in Adults: Guidelines for Management. Clinical Infectious Diseases 1998;26:811–38. [PubMed]
5. Bonilla F, Bernstein IL, Khan D, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol 2005; 94:S1-63. [PubMed]
6. Boyum A, Wiik P, Gustavsson E, et al. The effect of strenuous exercise, calorie deficiency and sleep deprivation on white blood cells, plasma immunoglobulins and cytokines. Scand J Immunol 1996; 43:228-235. [PubMed]
7. Buckley RH. Primary cellular immunodeficiencies. J Allergy Clin Immunol 2002; 109:747-757. [PubMed]
8. Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immunoglobulin in the prevention of pneumonia in patients with common variable immunodeficiency. J Allergy Clin Immunol 2002; 109:1001-1004.[PubMed]
9. Castellano G, Moreno D, Galvao O, et al. Malignant lymphoma of jejunum with common variable hypogammaglobulinemia and diffuse nodular hyperplasia of the small intestine. A case study and literature review. J Clin Gastroenterol 1992;15:128-135. [PubMed]
10. CDC. USPHS/IDSA guidelines for the prevention of opportunistic infections in persons infected with human immunodeficiency virus. MMWR 2002;51(No. RR-08):1–46.
11.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol 1999; 92:34-48. [PubMed]
12. Cunningham-Rundles C. Hematologic complications of primary immune deficiencies. Blood Reviews 2002; 16:61-64. [PubMed]
13. Davis AEI. C1 inhibitor gene and hereditary angioedema. In: Volanakis JE, Frank MM, editors. The human complement system in health and disease. New York: Marcel Dekker 1998; 229-244. [PubMed]
14. De Marie, S. Diseases and drug-related interventions affecting host defense. Eur J Clin Microbiol Infect Dis 1993; 12:S36. [PubMed]
15. Diagnosis and Management of Acute Otitis Media. American Academy of Pediatrics Subcommittee on Management of Acute Otitis Media.Pediatrics. 2004;113:1451-1465. [PubMed]
16. Eibl M. Immunological consequences of splenectomy. Prog Pediatr Surg 1985; 18:139. [PubMed]
17. Faist E. Schinkel C. Zimmer S. Update on the mechanisms of immune suppression of injury and immune modulation. World J Surg 1996; 20:454. [PubMed]
18. Farr M, Tunn E, Bacon PA, Smith DH. Hypogammaglobulinemia and thrombocytopenia associated with sulphasalazine therapy in rheumatoid arthritis. Ann Rheum Dis 1985; 44:723-724. [PubMed]
19. Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev 1991; 4:359-95. [PubMed]
20. Folds JD, Schmitz JL. Clinical and laboratory assessment of immunity. J Allergy Clin Immunol 2003; 111:S702-S711. [PubMed]
21. Fuss U, Strober W, Cuccherini BA, et al. Intestinal lymphangiectasia, a disease characterized by selective loss of naive CD45RA+ lymphocytes into the gastrointestinal tract. Eur J Immunol 1998; 28:4275-4285. [PubMed]
22. Glaser R, Rice J, Sheridan J, et al. Stress-related immune suppression: health implications. Brain Behav Immun 1987; 1:7. [PubMed]
23. Hamilos DL, Young RM, Peter JB, Agopian MS, Ikle DN, Barka N. Hypogammaglobulinemia in asthmatic patients. Ann Allergy 1992; 68:472-481. [PubMed]
24. Iliopoulos AG, Tsokos GC. Immunopathogenesis and spectrum of infections in systemic lupus erythematosus. Semin Arthritis Rheum 1996; 25:318. [PubMed]
25. Irwin M. Stress induced immune suppression: Role of brain corticotropin releasing hormone and autonomic nervous system mechanisms. Adv Neuroimmunol 1994; 4:29. [PubMed]
26. Jenkins C, Costello J, Hodge L. Systematic review of prevalence of aspirin induced asthma and its implications for clinical practice. BMJ 2004; 328:434. [PubMed]
27. Knight AK, Cunningham-Rundles C. Inflammatory and autoimmune complications of common variable immune deficiency. Autoimmunity Reviews 5. Elsevier BV 2006; 156-159.
28. Kuo MC, Hwang SJ, Chang JM, Tsai JC, Tsai JH, Lai YH. Recurrent infections in haemodialysis patients - do not forget selective immunoglobulin A deficiency. Nephrol Dial Transplant 1998; 13:3220-3222. [PubMed]
29. Leibovici L. Paul M. Cullen M. Bucaneve G. Gafter-Gvili A. Fraser A. Kern WV. Antibiotic prophylaxis in neutropenic patients: new evidence, practical decisions. Cancer. 2006;107:1743-51. [PubMed]
30. Leinonen M, Sakkinen A, Kallikoski R, et al. Antibody response to 14-valent pneumococcal capsular polysaccharide vaccine in pre-school age children. Pediatr Infect Dis 1986;5:39-44. [PubMed]
31. Lo N. Cullen M. Antibiotic prophylaxis in chemotherapy-induced neutropenia: time to reconsider. Hematological Oncology 2006;24:120-5. [PubMed]
32. Meeks M, Bush A. Primary ciliary dyskinesia (PCD). Pediatr Pulmonol 2000; 29:307-316. [PubMed]
33. Michel M, Chanet V, Galicier L, et al. Autoimmune thrombocytopenic pupura and common variable immunodeficiency: analysis of 21 cases and review of the literature. Medicine 2004; 83:254-263. [PubMed]
34. Monto, AS. Viral respiratory infections in the community: Epidemiology, agents and interventions. Am J Med 1995; 99:24S. [PubMed]
35. Moreno-Ancillo A, Cosmes Martin PM, Dominquez-Noche C, et al. Carbamazepine induced transient monoclonal gammopathy and immunodeficiency. Allergol Immunopathol (Madr) 2004; 32:86-88. [PubMed]
36. Noroski LM, Shearer WT. Screening for primary immunodeficiencies in the clinical immunology laboratory. Clin Immunol Immunopathol 1998; 86:237-245. [PubMed]
37. Ogi M, Yokoyama H, Tomosugi N, et al. Risk factors for infection and immunoglobulin replacement therapy in adult nephrotic syndrome. Am J Kidney Dis 1994; 24:427-436. [PubMed]
38. Orange JS, Hossny EM, Weiler CR, et al. Use of intravenous immunoglobulin in human disease: A review of evidence by members of the Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol 2006;117:S525-548. [PubMed]
39. Perri RT, Oken MM, Kay NE. Enhanced T cell suppression is directed toward sensitive circulating B cells in multiple myeloma. J Lab Clin Med 1982; 99:512-519. [PubMed]
40. Petri M. Infections in systemic lupus erythematosus. Rheum Dis Clin North Am 1998; 24:423. [PubMed]
41. Pettit SJ, Bourne H, Spickett GP. Survey of infection in patients receiving antibody replacement treatment for immune deficiency. J Clin Pathol 2002; 55:577-580. [PubMed]
42. Pickering M, Botto M, Taylor RP, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol 2000; 76:227-324. [PubMed]
43. Rachelefsky GS, Katz RM, Siegel SC: Chronic sinus disease with associated reactive airway disease in children. Pediatrics 1984; 74:526-529. [PubMed]
44. Rose M, Lang D. Evaluating and managing hypogammaglobulinemia. Cleve Clin J Med 2006; 73(2):133-144. [PubMed]
45. Santos JI. Nutrition, infection, and immunocompetence. Infect Di Clin North Am 1994; 8:243. [PubMed]
46. Schulze DH, Goidl E. Age associated changes in antibody forming cells (B cells). Proc Soc Exp Biol Med 1991; 99A:1. [PubMed]
47. Sekul EA, Cupler EJ, Dalakas MC. Aseptic meningitis associated with high dose intravenous immunoglobulin: frequency and risk factors. Ann Intern Med 1994; 121:259-262. [PubMed]
48. Sewell WC, Bucklana MS, Jolles SR. Therapeutic strategies in common variable immunodeficiency. Drugs 2003; 63:1359-1371. [PubMed]
49. Slavin R. Spector S. Bernstein L. The Diagnosis and Management of Sinusitis: A Practice Parameter Update. JACI. 2005;116:S13-47. [PubMed]
50. Snowden N, Dietch DM, Teh LS, Hilton RC, Haeney MR. Antibody deficiency associated with gold treatment: natural history and management in 22 patients. Ann Rheum Dis 1996; 55:616-621. [PubMed]
51. Stiehm ER. Human intravenous immunoglobulin in primary and secondary antibody deficiencies. Pediatr Infect Dis J 1997; 16:696-707. [PubMed]
52. Stiehm ER, Ochs HD, Winkelstein JA. Immunodeficiency disorders: general considerations. In: Stiehm ER, Ochs HD, Winkelstein JA, eds. Immunologic Disorders in Infants & Children. 5th ed. London, England: Elsevier Saunders; 2004:289-355.
53. Stray-Pedersen A, Abrahamsen TG, Froland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol 2000; 20:477-485. [PubMed]
54. Travin M, Macris NT, Block JM, Schwimmer D. Reversible common variable immunodeficiency syndrome induced by phenytoin. Arch Intern Med 1989; 149:1421-1422. [PubMed]
55. Tunkel A. Et al. Practice Guidelines for the Management of Bacterial Meningitis. Clinical Infectious Diseases 2004; 39:1267–84. [PubMed]
56. Uluhan A, Sager D, Jasin HE. Juvenile rheumatoid arthritis and common variable hypogammaglobulinemia. J Rheumatol 1998; 25:1205-1210. [PubMed]
57. Von Andrian UH, Mackay CR. T-cell function and migration. NEJM 2000; 343:1020-1034. [PubMed]
58. Wen L, Atkinson J, Giclas P. Clinical and laboratory evaluation of complement deficiency. J Allergy Clin Immunol 2004; 113:585-593. [PubMed]
59. Yarmohammadi H, Estrella L, Doucette J, Cunningham-Rundles C. Recognizing Primary Immune Deficiency in Clinical Practice. Clin and Vaccine Immunol 2006; 13:329-332. [PubMed]
60. Yesliova Z, Ozata M, Kocar IH, et al. The effects of gonadotropin treatment on the immunological features of male patients with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2000; 85:66-70. [PubMed]
61. Zelazko M, Carneiro-Sampeio M, Cornejo de Luigi M, et al. Primary immunodeficiency diseases in Latin America: first report from eight countries participating in the LAGID (Latin American Group for Primary Immunodeficiencies). J Clin Immunol 1998; 18:161-166. [PubMed]
Tables
Table 1: Guidelines for Identifying Patients with Increased Susceptibility to Infections and a Possible Impaired Immune Response
| Frequency | Acute sinusitis 3 or more times per year (with objective evidence such as changes on computed tomography (CT) of the sinuses or pus visualized draining from a sinus ostium on rhinoscopy) |
| Pneumonia 2 or more times per year (documented with chest x-ray or CT scan findings) | |
| Severity | Pneumonia with empyema or necrotizing granuloma formation |
| Bacterial meningitis, arthritis, or osteomyelitis | |
| Sepsis | |
| Infection with Opportunistic Pathogens | |
| Pneumocystis carinii pneumonia | |
| Mucocutaneous candidiasis | |
| Invasive fungal infection | |
Table 2: Examples of Known Primary Immune Deficiencies
| Humoral Immunodeficiency | X-linked agammaglobulinemia (Bruton’s) |
| Common variable immunodeficiency | |
| Selective IgA deficiency | |
| Specific antibody deficiency | |
| Transient hypogammaglobulinemia of infancy | |
| Cellular Immunodeficiency | IL-12 defects |
| Interferon-gamma defects | |
| Chronic mucocutaneous candidiasis | |
| Combined Humoral and Cellular Immunodeficiency | |
| Severe combined immunodeficiency (SCID) | |
| Wiskott-Aldrich syndrome | |
| Ataxia-telengiectasia | |
| Hyper-IgM syndrome | |
| X-linked lymphoproliferative syndrome | |
| Phagocytic Cell Disorders | Chronic granulomatous disease |
| Chediak-Higashi syndrome | |
| Leukocyte adhesion deficiency | |
| Hyper-IgE syndrome (Job’s) | |
| Complement Deficiency | |
Table 3: Overview of how best to properly identify possible sinusitis, pneumonia, otitis media, or meningitis in a patient based upon presentation and proper imaging and laboratory data.
| Anatomic Location | Signs & Symptoms | Imaging Studies | Laboratory Data |
|---|---|---|---|
Tenderness overlying sinuses Dark circles beneath eyes Periorbital edema Pharyngeal erythema Purulent material in posterior pharynx Nasal mucosal erythema Purulent material inside nose Facial pain Low grade fever > 100.5 F Tooth pain Nasal congestion; particularly when unilateral or worse on one side Postnasal drip Cough (worse when supine) **All symptoms are non specific for sinusitis and are not diagnostic of bacterial sinusitis |
May not need any imaging for acute sinusitis (symptoms < 4 weeks)
Chronic (>8 wks or failed medical therapy in acute sinusitis) or Recurrent sinusitis (>3 episodes/year): Simple coronal sinus CT scan without contrast -Mucosal thickening > 6mm in maxillary sinuses OR - > 33% loss of air space volume within maxillary sinuses OR -Opacification-air-fluid levels in any paranasal sinus -Evaluate for anatomic abnormalities which may predispose to infection
Plain radiographs offer suboptimal sensitivity and specificity
|
Nasal cultures are not reliable
Maxillary antrum aspiration provides definitive culture but is only indicated when precise identification is essential
Cultures obtained through middle meatus via anterior rhinoscopy are helpful when available
Consider evaluation of Humoral Immune System for recurrent sinusitis |
|
Pneumonia4 |
Cough Sputum production Dyspnea Fever Altered breath sounds |
Anterior-posterior and lateral plain chest radiograph -Opacification of one or more lobes *CXR findings may lag behind clinical presentation
Chest CT scan indicated for recurrent pneumonia to evaluate for possible anatomic abnormalities which may predispose to infection -Bronchiectasis
Consider bronchoscopy for recurrent pneumonia not responsive to antibiotics |
Deep cough expectorated sputum sample -Gram stain -Culture *Sample should be transported to lab within 3-4 hours of collection
Blood culture
CBC with differential
Consider evaluation of Humoral Immune System for recurrent pneumonia
|
Otitis Media15 |
Otalgia Otorrhea Fever **Symptoms have poor correlation with actual bacterial infection |
No imaging studies are necessary, rely on physical exam
Tympanic membrane may show: -Bulging and fullness best predictors of acute otitis media -Redness -Diminished light reflex -Pneumatic otoscopy is important to determine decreased movement of TM |
Tympanocentesis provides definitive culture but is only indicated when precise identification is essential
Consider evaluation of Humoral Immune System for recurrent otitis media -Evaluate closely for co-morbid conditions which may predispose to recurrent otitis |
Fever Neck pain/stiffness Seizures Focal neurological deficits Purpuric rash (Neisseria) |
Consider head CT prior to lumbar puncture in certain patients (see referenced guidelines for complete list) |
Lumbar puncture -Gram stain -Culture -Elevated WBC (left shift) -Decreased Glucose -Elevated protein
Blood culture
Consider evaluation of terminal Complement System (C5-C9) for a patient with recurrent Neisseriainfections
Consider evaluation of Humoral Immune System for recurrent bacterial/enteroviral meningitis |
Table 4: Primary Immunodeficiency and Associated Pathogens:
Immune defect/dysfunction Associated Pathogens
Humoral (Antibody) Immune System |
Pyogenic encapsulated bacteria: Gram negative bacteria:
|
Cellular (T cell mediated) Immune System |
Viruses:
Mycobacteria:
Fungi: |
Phagocytic Immune System |
Catalase producing bacteria: Salmonella
Fungi: Candida |
Complement System |
Deficiency in early complement proteins: Pyogenic encapsulated bacteria
Deficiency in late complement proteins: Also consider in a patient with connective tissue disease, e.g. systemic lupus erythematosus |
Table 5: Normal Serum Immunoglobulin Concentrations
Immunoglobulin type |
Normal values in adults (mg/dL)
|
|---|---|
IgG |
717-1,411 |
IgG1 |
344-966 |
IgG2 |
133-622 |
IgG3 |
12-138 |
IgG4 |
1-115 |
IgA |
78-391 |
IgM |
3-334 |
Table 6: FDA Approved Indications for IVIG
1. Primary immunodeficiency disease or primary humoral immunodeficiency
2. Idiopathic thrombocytopenic purpura
3. Kawasaki disease (syndrome)
4. B-cell chronic lymphocytic leukemia
5. HIV infection (pediatric patients)
6. Bone marrow transplantation (patients >20 years of age in first 100 days after transplant)